Breaking the Code
- LAC
- Highlights
The group of Prof. Copéret and an international team of researchers show how NMR spectroscopy can be used to understand and predict the electronic structure and reactivity of molecules by decoding the information contained in the NMR chemical shift.

Understanding the relation between structure and reactivity for catalytic intermediates in chemical reactions is key for the development of more sustainable processes. Studying these molecules by spectroscopic techniques provides the basis for understanding, rationalizing and designing catalytic reactions. Among these techniques, Nuclear Magnetic Resonance (NMR) spectroscopy has emerged as one of the most powerful tools to monitor and understand the structure of catalysts and catalytic intermediates. NMR is very sensitive to the local bonding and environment of the atoms that constitute molecular structures. In NMR spectroscopy, molecules are exposed to a very high magnetic field (around 10-20 Tesla, which is almost 1 million times stronger than the earth magnetic field); this induces local magnetic fields in the molecule, which are slightly different from atom to atom. This property is called chemical shift and it is measurable for any atom with a nuclear spin. The values are reported in ppm (parts per million shifts as compared to the large external magnetic field). These chemical shift values, which are extremely sensitive to the local environment of each atom, are often referred to as spectroscopic signatures, since different molecules show different and very specific NMR spectra (ensemble of signals). Overall, NMR spectroscopy thus provides insights about the structure of molecules (how atoms are bonded to each other) but also into the distribution of electrons around the nuclei. While chemists use NMR (and chemical shift values) to characterize molecules and chemical reactions, the actual chemical shift values are often regarded as mere numbers and only used empirically to recognize a structure.
An international team of researchers from ETH Zürich, UC Berkeley, Université de Montpellier and the University of Oslo has investigated what is behind these chemical shift values in a series of similar molecules and gained unprecedented insights, which allow connecting chemical shift values to their catalytic activity in olefin metathesis. Olefin metathesis is a catalytic chemical reaction that has significantly impacted chemistry in recent years, both in industry and in academia. In this reaction, two carbon-carbon double bonds are cleaved and reassembled, thus generating new molecules in a very efficient way (minimizing energy and byproduct formation). The development of olefin metathesis catalysts was hence awarded a Nobel Prize in 2005. This catalytic reaction involves metal-carbenes (molecules with a metal-carbon double bond) and metallacyclobutanes (cyclic structures consisting of one metal atom and three carbon atoms) as key reaction intermediates that allow the exchange of carbon-carbon double bonds among olefins (Scheme 1). These species – metal carbenes and metallacyclobutanes – are associated with specific chemical shifts. In particular, it was observed over the years that metallacyclobutanes that could participate in olefin metathesis were associated with characteristic carbon-13 chemical shift values (around 100 ppm and 0 ppm), while metallacyclobutanes unable to participate in olefin metathesis had very different chemical shifts (30-40 ppm). These specific chemical shifts have puzzled the scientific community involved in the development of these catalysts for decades: while there seemed to be a link between the specific chemical shift values and the reactivity of the metallacyclobutane, it was not clear what was their origin and whether or not a secret information was encoded in these values.
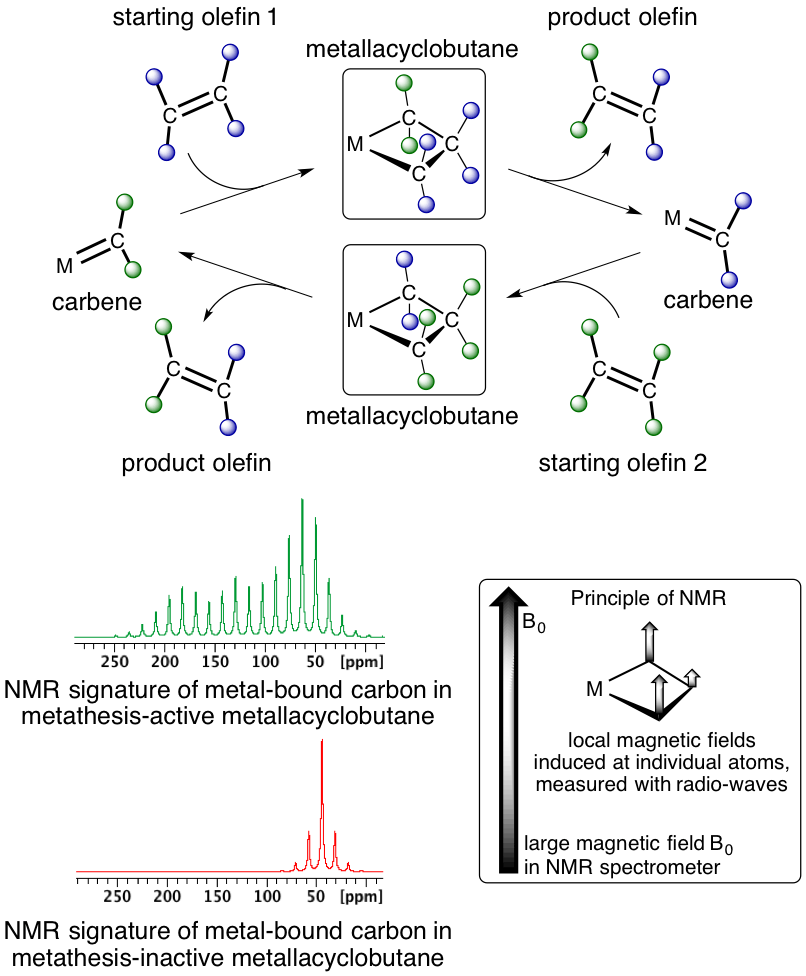
The team of researchers studied the quantum chemical origin of these chemical shift values by a combination of NMR spectroscopy and computational chemistry; such detailed analysis became possible recently because of the developments of more powerful computers and related quantum chemical analytical tools. In this study, it was found that the very distinct NMR-spectroscopic signatures for the metallacyclobutanes originated from a specific electronic structure of the reactive intermediate in olefin metathesis. In particular, these studies have shown that the carbon attached to the metal in the reactive metallacyclobutane contains some remaining carbene character (coming from the original reacting partner, the metal-carbene) to facilitate the exchange of the carbon fragments by metathesis. In contrast, other metallacyclobutanes do not contain any information related to carbenes and are thus unreactive.
This work connects for the first time the reactivity of molecules to values that can be easily accessible by NMR spectroscopy. In other words, this study has helped to decode the reactivity of molecules by deciphering the origin of readily accessible chemical shift values. This approach constitutes a step towards the predictive understanding of the reactivity of catalysts and molecules by decoding their NMR signatures, a first step towards the rational design of sustainable catalytic processes.
This work was published recently in ACS Central Science, entitled “Metathesis Activity Encoded in the Metallacyclobutane Carbon-13 NMR Chemical Shift Tensors”.
References
C. P. Gordon, K. Yamamoto, W.-C. Liao, F. Allouche, R. A. Andersen, C. Copéret, C. Raynaud, O. Eisenstein
external pageMetathesis Activity Encoded in the Metallacyclobutane Carbon-13 NMR Chemical Shift Tensors, ACS Central Science, 2017, DOI: 10.1021/ACSCENTSCI.7B00174.
external pageLiveslides explaining the publication, presented by Christopher Gordon